An important update is available for FreeStyle LibreLinkØ. Check here for more information.
If you know the product code enter it here

The SMART-Seq Stranded Kit is used to generate strand-specific RNA-seq libraries for Illumina® sequencing from 1–1,000 sorted cells or 10 pg–10 ng of purified total RNA. This kit incorporates Takara Bio's SMART (Switching Mechanism at the 5' end of RNA Template) technology and includes refinements to the SMARTer method for stranded RNA-seq that simplify the library preparation workflow and improve sequencing performance.
Brand: Takara
Go to checkout
The SMART-Seq Stranded Kit is used to generate strand-specific RNA-seq libraries for Illumina® sequencing from 1–1,000 sorted cells or 10 pg–10 ng of purified total RNA. This kit incorporates Takara Bio's SMART (Switching Mechanism at the 5' end of RNA Template) technology and includes refinements to the SMARTer method for stranded RNA-seq that simplify the library preparation workflow and improve sequencing performance. In addition, contrary to the SMART-Seq v4 Ultra Low Input RNA Kit for Sequencing and SMART-Seq HT Kit, reverse transcription is initiated using random priming instead of oligo(dT) priming, thus capturing the full transcriptome instead of only the polyadenylated fraction. The SMART-Seq Stranded Kit was specifically designed to deliver highly sensitive and reproducible data from single cells while keeping the workflow short and user friendly. The kit does not require additional rRNA removal methods or kits and produces sequencing libraries that retain strand-of-origin information. The integrated removal of cDNAs derived from rRNA-typically present in high abundance following cDNA synthesis from total RNA inputs-makes the workflow extremely sensitive, yielding data that is highly reproducible with low mapping to rRNA.
Interested in more data and FAQs about this product? Visit the NGS Learning Center.
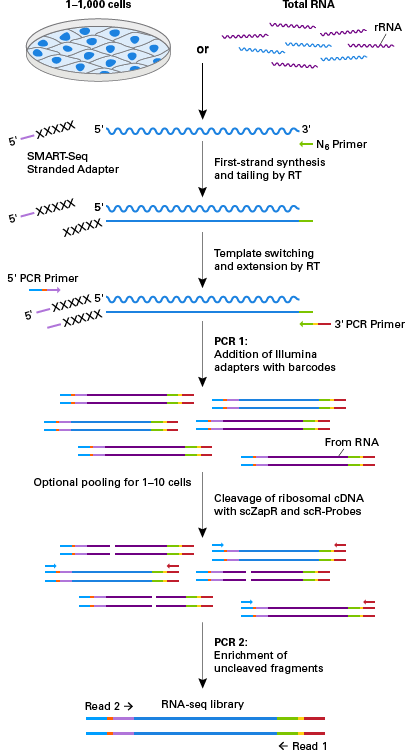
Figure 1. Schematic of the SMART-Seq Stranded Kit workflow.
| Sequencing alignment metrics for A375 total RNA and cells | |||||||
| Input | Total RNA | 1,000 cells | 500 cells | 100 cells | 10 cells | 5 cells | 1 cell |
| Number of reads (pairs) | 6,000,000 | 6,000,000 | 6,000,000 | 6,000,000 | 6,000,000 | 6,000,000 | 5,873,974 |
| Number of transcripts >1 FPKM | 13,260 | 13,294 | 13,583 | 13,520 | 12,726 | 12,602 | 11,540 |
| Number of transcripts >0.1 FPKM | 21,334 | 21,113 | 21,365 | 21,145 | 20,550 | 18,888 | 15,815 |
| Proportion of reads (%): | |||||||
| Exonic | 34.7 | 36.4 | 39.2 | 42.7 | 36.7 | 36.2 | 37.3 |
| Intronic | 29.6 | 29.3 | 27.7 | 28.3 | 34.0 | 30.4 | 21.1 |
| Intergenic | 14.2 | 13.4 | 12.2 | 12.9 | 16.7 | 16.8 | 10.1 |
| rRNA | 7.0 | 11.4 | 11.5 | 6.3 | 3.6 | 4.9 | 7.1 |
| Mitochondrial | 4.1 | 3.5 | 3.7 | 4.9 | 3.8 | 4.4 | 4.6 |
| Overall mapping (%) | 89.6 | 93.9 | 94.3 | 95.1 | 94.9 | 92.7 | 80.2 |
| Duplicate rate (%) | 37.3 | 45.2 | 40.3 | 46.1 | 52.5 | 72.2 | 78.5 |
| lncRNA mapping: | |||||||
| Number of mapped reads (%) | 7.2 | 10.4 | 10.8 | 9.4 | 8.7 | 8.6 | 7.3 |
| lncRNA transcripts detected | 5,395 | 4,687 | 4,565 | 5,439 | 5,440 | 4,983 | 2,802 |
Figure 2: High reproducibility across cell input amounts. A375 cells isolated by FACS were used to generate RNA-seq libraries with the SMART-Seq Stranded Kit. Input varied from 1 cell to 1,000 cells, with two replicates per input of 5–1,000 cells and 12 replicates for the single cells. For comparison, two aliquots of 1,000 cells were used for total RNA purification and then used for library preparation.
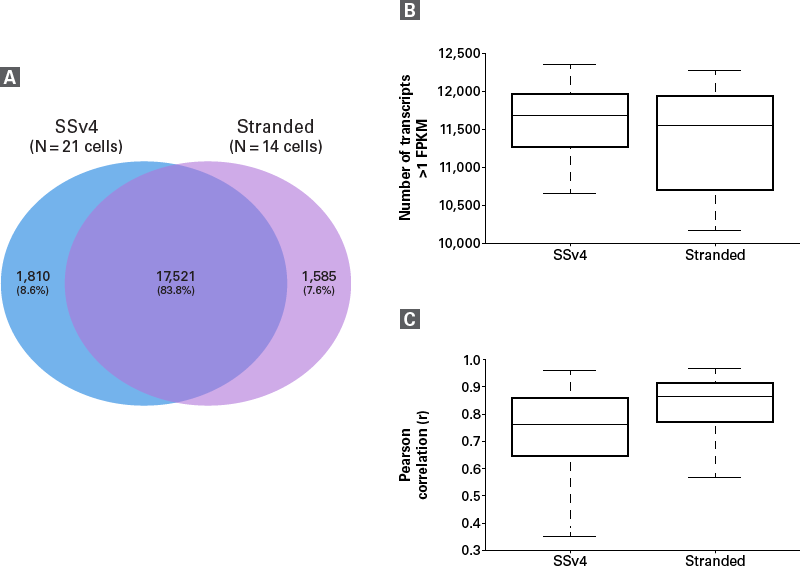
Figure 3. Comparison between SMART-Seq v4 and SMART-Seq Stranded kits. Single cells (K562) isolated by FACS were used to generate RNA-seq libraries with the SMART-Seq Stranded Kit (Stranded) and a SMART-Seq v4 Kit (SSv4; cDNA from this kit was further processed with a Nextera® XT DNA Library Preparation Kit). Panel A. The overlap in the total number of transcripts identified (FPKM >1) by each kit was analyzed and shown to be 83.8%. Panel B. The number of transcripts identified (FPKM >1) in individual cells was similar between the two kits, with a tighter range across cells processed with the SSv4 kit. Panel C. The reproducibility (Pearson correlation) of transcript expression levels across all cells from each kit was similar, although slightly higher and more consistent across cells processed with the Stranded kit.


