An important update is available for FreeStyle LibreLinkØ. Check here for more information.
27 Sep 2018

Whether you are performing a single parameter, simple flow cytometry experiment, or a 17 colour multiparameter experiment, there should always be a common set of best practices to follow, just like any scientific experiment. As the complexity increases, so too does the necessity for controls, optimisation, and validation. Missing just one control can mean the waste of a whole experiment, or worse, a manuscript getting rejected.
There are a number of important controls to consider when designing your experiments:
There are a number of controls that can be considered staining controls – i.e. those controls that help validate, or confirm that the fluorescent signals that you measure are an accurate reflection of the biology. The first aspect to consider is whether antibody binding is indeed specific – i.e. that antibodies are correctly binding (antigen-specifically) to their target and not off-target or randomly (non-specifically).
1. Controlling for non-specific binding:
Sources of off-target or unwanted, non-specific binding:
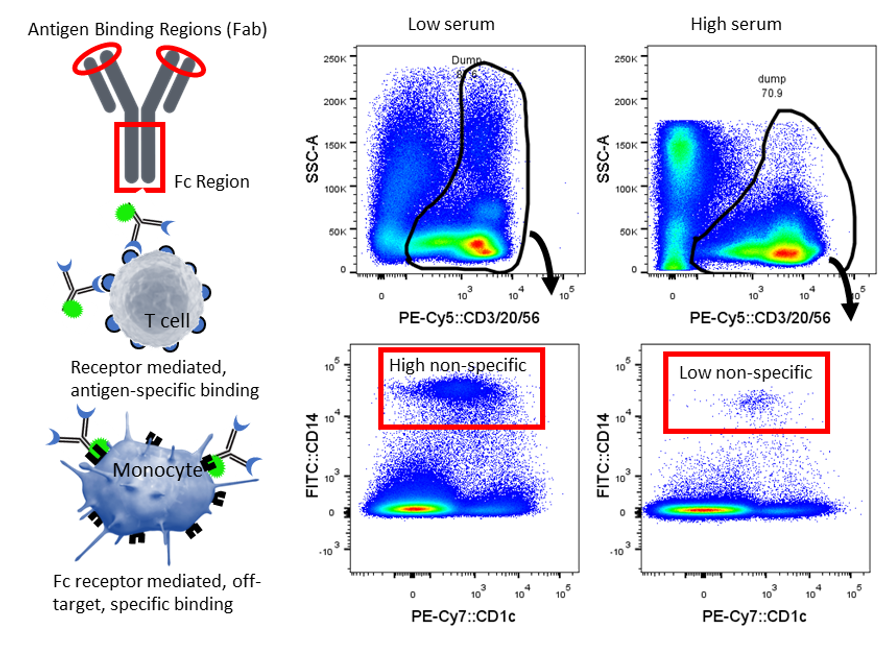
This figure demonstrates that sometimes antibodies can stick non-specifically – or more accurately, specifically, but off-target. Cells with Fc receptors such as monocytes and macrophages can bind the Fc portion of an antibody, resulting in a false-positive signal. Antibody specificity is via the Fab region and so this unwanted Fc binding interaction is best mitigated by using blocking reagents or serum. Monocytes (CD14+) non-specifically bind the dump channel cocktail (CD3/20/56) either via Fc binding, or by recognition of PECy5. This non-specific binding is reduced when staining is performed in whole blood (i.e. high serum) compared to PBMC prepared in staining buffer (1%FCS). Image courtesy of Dr Anna Brooks.
What about Isotype controls?
Isotype controls are meant to be a control for non-specific binding, however, it is often difficult to correctly match the reagent in question. To correctly use an isotype control, the fluorophore to protein (F/P) ratio should be the same. In addition, the isotype control should be raised in the same host species against an irrelevant antigen of the same Ig subclass, with the same conjugated fluorophore that is obtained from the same supplier as your test antibody and used at the same concentration. However, this information may not always be obtainable or obvious. Indeed if all conditions are met, then an isotype control may help demonstrate specificity, or lack thereof. These days, isotype controls are largely considered “old-school” or obsolete, not because they have become unfashionable, but because they are not a particularly good control (Maecker et al 2006). In some instances, it can be tempting to at least control for non-specific staining, i.e. intracellular staining, or Fc receptor binding, but in reality, isotype controls don't work perfectly. Therefore, prevention is the best method and is achieved by Fc blocking, use of viability stains and properly titrating reagents.
Titrating reagents
There is a misconception that titrating antibodies is just a money-saving activity (that many choose to avoid), however using excess antibody for intracellular applications or large multicolour panels can actually prevent accurate resolution. Multicolour panels rely heavily on using optimal concentrations of antibody. Excessively high concentrations can reduce signal-to-noise ratio and increase non-specific binding. Excessively low concentrations can result in insufficient staining to resolve dim populations or weak expression. Where possible, titrate all antibodies using the tissue of interest, as well as the staining conditions (i.e. antibody incubation temperature and duration). Sometimes, a small panel of antibodies may be required to “drill down” or exclude cells in order to get an accurate assessment of antibody dose on cells with a low frequency. This is fairly common for large multicolour panels of complex tissue, where many target cell populations are at a low frequency.
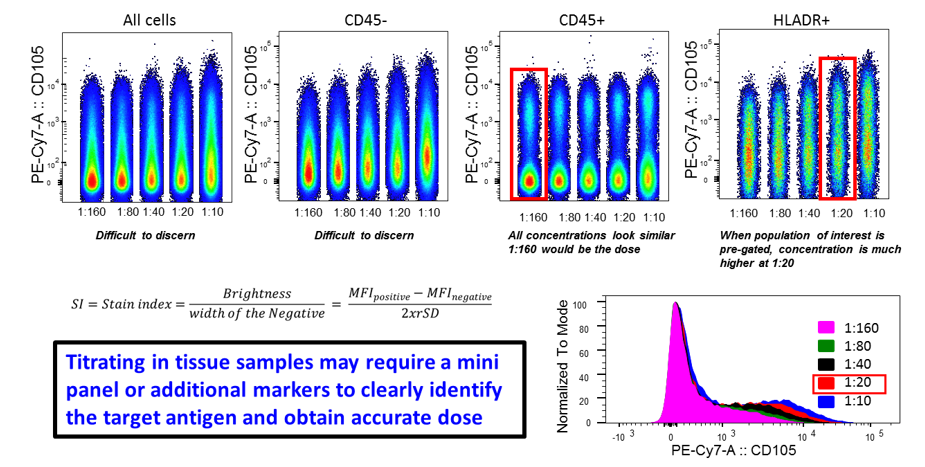
This figure demonstrates that titrations can be difficult to interpret when multiple cell types express the same antigen at different levels in a complex sample. It can be a good idea to use a mini panel to “drill-down” to the cells of interest in order to get an accurate dose. When assessing all CD45+ cells a dose of 1:160 appeared sufficient. However when the population of interest, an HLADR+ population was assessed, the best dose was higher, at 1:20. Image courtesy of Dr Anna Brooks.
2. Viability/dead cell exclusion dyes
An important control in any flow cytometry experiment is to confirm that the cells being interrogated are indeed viable, or more importantly to exclude dead cells that non-specifically bind antibody. There are a number of types of dyes that can be used, depending on the application.
|
|
Target/ binds to |
Cell permeant |
Cell semi-permeant or impermeant |
Intact cell marker |
Viability marker |
Dead marker |
Fixable |
|
Live/Dead fixable |
Amine reactive |
Y |
|
Y |
Y |
||
|
Amine reactive |
Y |
|
Y |
Y |
|||
|
Helix NP™ |
dsDNA |
Y |
|
Y |
N |
||
|
7AAD |
DNA/RNA |
Y |
|
Y |
N |
||
|
Propidium Iodide PI |
DNA/RNA |
Y |
|
Y |
N |
||
|
DRAQ7™ |
dsDNA |
Y |
|
Y |
|||
|
DAPI |
dsDNA |
Y |
|
Y |
|||
|
Calcein dyes |
nonspecific esterases |
Y |
Y |
Y |
|
N |
|
|
CytoPhase™ Violet |
DNA |
Y |
|
Y |
|
|
Y |
|
Hoechst dyes |
dsDNA |
Y |
Y |
|
N |
||
|
SYTO™ blue/green/orange/red |
DNA/RNA |
Y |
Y |
Y |
N |
||
|
DRAQ5™ |
dsDNA |
Y |
Y |
N |
Cell permeant dyes: These dyes penetrate into all cells and are useful for identifying “intact” or real cell events among a “sea of noise”. This can be a useful way to identify cells in digested tissue samples that may have a lot of debris and extracellular matrix. For example, Calcein dyes (Calcein AM, Violet AM, and Calcein Blue AM) are esterase substrates which are activated to fluoresce once they cross the cell membrane and therefore selectively label live cells. Apoptotic and dead cells with compromised cell membranes, however, do not retain these dyes. Cellular debris can be a sticky mess and contribute to non-specific binding, so identifying real cell events with a permeant dye may be useful for this reason. Hoechst 33342 (a nucleic acid stain), by comparison, is useful for identifying intact live cells for sorting applications. However, it cannot be used for viability as it diffuses across the membranes of all cells.
Cell impermeant dyes: Nucleic acid dyes can be used to identify dead cells. These are a unique class of dye that are non-fluorescent until they undergo conformational changes upon binding to nucleic acid targets. Live cells exclude membrane impermeant dyes, but dead cells start to lose membrane permeability allowing dyes to gain access to intracellular components such as RNA/DNA when the plasma membrane is damaged. Therefore, cells with compromised membranes will stain brightly as the dye gains access to the intracellular targets. Examples include DAPI, 7AAD and PI.
Live/Dead fixable: These dyes come in a wide range of multiple single-colour formats and are compatible with multiple lasers making them ideal for dead cell exclusion in multicolour panels. These dyes covalently bind extracellular and intracellular amines. Live cells will also stain, but only weakly, whereas cells with compromised membranes stain brightly as the dyes gain access to intracellular proteins, staining them irreversibly. These dyes are compatible with protocols that require cryopreservation, fixation, and/or permeabilisation procedures without dead cell staining intensity loss. The Zombie Fixable ViabilityTM dyes from BioLegend are an example of these dyes and come in a wide range of colour options.
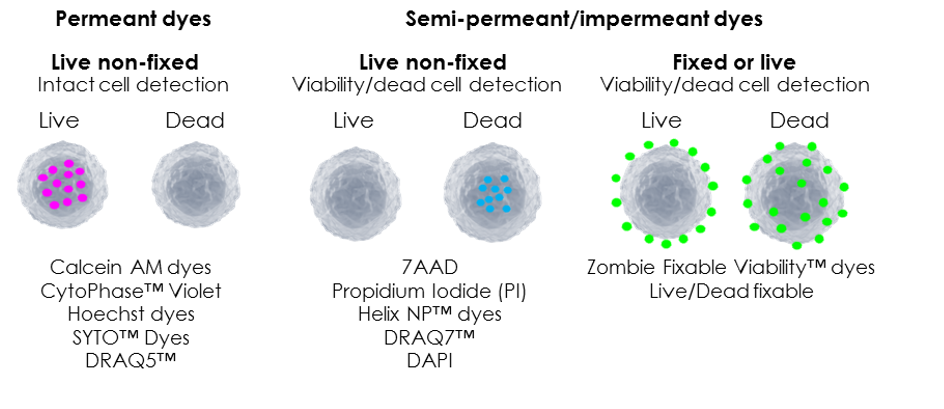
Image courtesy of Dr Anna Brooks.
3. Reference sample or “normal” controls
When performing longitudinal studies, it is often a good idea to use a reference sample (e.g. a healthy donor, or untreated mice). This sample is then treated in the exact same manner for every experiment within a study (e.g. thawed, fixed or treated) so that if any deviation from the norm occurs, it will be flagged as a technical problem rather than mistaken for a biological observation.
Dr Anna Brooks holds a BCA (management) and a PhD in Immunology and is a Senior Research Fellow with the Maurice Wilkins Centre at the University of Auckland. Anna is also an expert flow cytometrist and is director of Auckland Cytometry, the flow cytometry core facility for the Faculty of Science. Anna’s primary interest lies in developing multicolour panels to characterise complex cellular populations in digested human tissues. Anna is also an active member of the international flow cytometry community and currently sits on the Australasian Cytometry Society Council as research liaison. When not marvelling in the many fluorescent colours of the cytometry world, Anna enjoys to scuba dive and explore the vibrant wonders of our underwater world.

Dr Anna Brooks
University Of Auckland, School Of Biological Sciences
27 Sep 2018
If you enjoyed reading our articles, why not sign up to our blog mailing list? You'll get new articles straight to your inbox as they're released!



